
Muscular Dystrophy and
the Cavalier King Charles Spaniel
-
 What It Is
What It Is - Symptoms
- Diagnosis
- DNA Testing
- Treatment
- Breeders' Responsibilities
- Research News
- Related Links
- Veterinary Resources
What It Is
Some cavalier King Charles spaniels suffer from a genetic form of muscular dystrophy (MD) caused by a mutation of the dystrophin gene. Dystrophin is a protein that acts to stabilize skeletal and cardiac muscles and provides mechanical stability during the muscles’ contractions. The dystrophin gene encodes that protein. The mutations in the dystrophin gene causes an absence of dystrophin in the muscles, which results in progressive degeneration of muscle fibres.
Muscular dystrophy is hereditary in the cavalier but is carried only by the X chromosome. Since males have only one X chromosome, they are more susceptible to inheriting MD because they need only one parent to pass the gene to them. For females to inherit the gene, both parents must carry it. Females are more often only carriers of the gene.
The genetic cause of muscular dystrophy in the CKCS was identified in a 2009 report by a research team at the UK Royal Veterinary College, led by Dr. Richard. J. Piercy and Gemma L. Walmsley. This study is on-going, with the cooperation of Dr. G. Diane Shelton at the Comparative Neuromuscular Laboratory. For example, in a December 2016 article, researchers applied "whole genome, next generation sequencing" (WGS) to evaluate a an MD-affected cavalier. WGS detected a deletion mutation in a DMD gene hotspot which is distinct from a previously identified mutation.
In a 2010 study, the same group of researchers have opined that all affected cavaliers may be related to a single founder. The extent of this disorder in the breed is not known. In a study examining 96 female CKCSs in the UK, there were no carriers identified. So, MD is suspected to be present at only a low level within the breed.
The type of MD in cavaliers is very similar to Duchenne muscular dystrophy (DMD) in humans. It reportedly afflicts one in 3,500 boys, and therefore it is one of the most common genetic disorders of children.
RETURN TO TOP
Symptoms
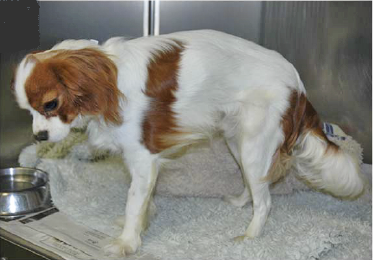
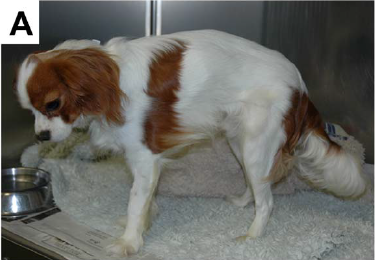
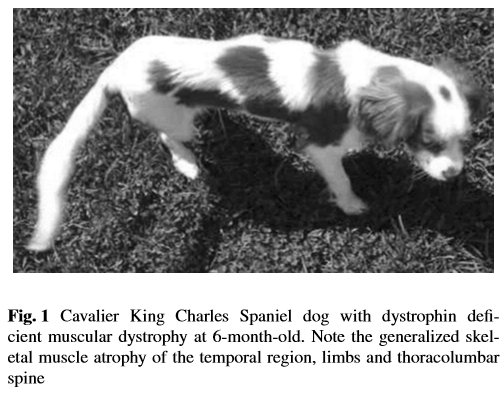
Since muscular dystrophy results in a lack of stability of the sketetal muscles, classic signs of MD are exercise intolerance, a stiff or hunched-up gait, and a lack of muscle mass. These symptoms usually are detectable at an early age, in some dogs, as young as six weeks to three months.
Other signs include: overall weakness, difficulty swallowing, an enlarged tongue, gagging when eating, breathing difficulties, muscle spasms, leg deformities, and tremors. The swallowing difficulty and gagging may result in aspiration pneumonia.
Symptoms in females usually are milder than in males.
Some mouth symptoms are similar to masticatory muscle myositis, another neuromuscular disease affecting the muscles used by the dog to chew – the jaw and temporal muscles.
RETURN TO TOP
Diagnosis
Based upon the dog’s symptoms, usually some sort of a neuromuscular disorder is suspected. The veterinarian should test the dog’s reflexes, tongue, and jaw movement.
A blood test would reveal that serum creatine kinase (CK) is highly elevated. A muscle biopsy, under general anesthesia, should be taken for evaluation. Immunohistochemistry analysis of the biopsy is an essential test to determine the absence of dystrophin in the muscles.
Research laboratories are able to determine whether cavaliers carry the mutation of the gene. UK's Animal Health Trust, with the Royal Veterinary College have identified the genetic mutation. VetNostic Laboratories Labogen, Laboklin, and MyDogDNA offer a test based on the mutation.
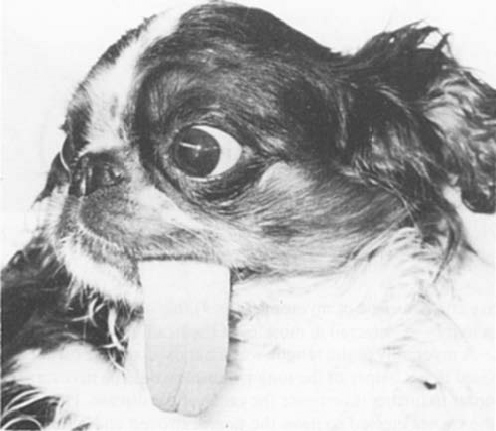 In a 1982 article about a
9-month-old female cavalier (right), she was unable to withdraw her tongue back
into her mouth. It was the only muscular disability she displayed. Upon
necropsy, all sections of the tongue showed replacement of muscle fibres
by adipose (fat) tissue. Paramyotonia, a type of
muscular dystrophy, was diagnosed. However, unlike "dystrophia
myotonica" paramyotonica is non-progressive and is not associated with
muscle wasting. Inheritance is in the pattern of a single autosomal
dominant and therefore unaffected members of families do not transmit
the disease. Also unlike myotonia congenita, paramyotonia usually occurs
only in the tongue, sometimes in the eyelids, and rarely in any other
muscle groups.
In a 1982 article about a
9-month-old female cavalier (right), she was unable to withdraw her tongue back
into her mouth. It was the only muscular disability she displayed. Upon
necropsy, all sections of the tongue showed replacement of muscle fibres
by adipose (fat) tissue. Paramyotonia, a type of
muscular dystrophy, was diagnosed. However, unlike "dystrophia
myotonica" paramyotonica is non-progressive and is not associated with
muscle wasting. Inheritance is in the pattern of a single autosomal
dominant and therefore unaffected members of families do not transmit
the disease. Also unlike myotonia congenita, paramyotonia usually occurs
only in the tongue, sometimes in the eyelids, and rarely in any other
muscle groups.
In a 2004 abstract, two cavaliers diagnosed with an absence of dystriphin also were found to have myoglobinuria (the presence of myoglobin in the urine, usually associated with muscle destruction) and myalgia (painful or tender muscles), which at that time were considered unusual in cases of canine MD. Myoglobinuria is described as dark ‘‘coca-cola’’ colored urine. A severe form of myoglobinuria is associated with massive muscle deterioration.
RETURN TO TOP
 DNA
Testing
DNA
Testing
Research laboratories are able to determine whether cavaliers carry the mutation of the dystrophin gene. UK's Animal Health Trust, with the Royal Veterinary College have identified the genetic mutation.
Genetic testing laboratories which test for this mutation causing CDM include:
• Embark, Cornell University College of Veterinary Medicine
• Laboklin
• MyDogDNA, Wisdom Panel
![]() Watch
this YouTube video to learn how to properly use a swab to obtain DNA from
your dog. See our Genetic Testing webpage for
more information about DNA testing.
Watch
this YouTube video to learn how to properly use a swab to obtain DNA from
your dog. See our Genetic Testing webpage for
more information about DNA testing.
RETURN TO TOP
Treatment
 Until
recently, there has been no cure for muscular dystrophy in the cavalier King Charles spaniel.
Nor has there been any medical therapy for maintenance of the disorder or to slow its
progression. An exception to this could be anabolic steroid supplements or
growth hormone treatments, which may temporarily slow the progression, but these
drugs have potentially damaging side effects and may not be used long term.
Until
recently, there has been no cure for muscular dystrophy in the cavalier King Charles spaniel.
Nor has there been any medical therapy for maintenance of the disorder or to slow its
progression. An exception to this could be anabolic steroid supplements or
growth hormone treatments, which may temporarily slow the progression, but these
drugs have potentially damaging side effects and may not be used long term.
Researchers have been examining the efficacy of transplanting stem cells into dogs with muscular dystrophy. One such research project has reported increases in the dogs’ level of dystrophin and regained muscle strength and mobility.
In an August 2018 article, researchers report that they tested the value of gene editing the deltaE50-MD (ΔEx50) mutated gene in four one-month old cavalier mix puppies (Bichon Frise x CKCS female mated with male Beagles). Two of the puppies were injected with adeno-associated viruses to deliver CRISPR gene editing components intra-muscularly and the other two were injected systemically in the cephalic vein. The four dogs were euthanized six to eight weeks later, and muscle tissue was collected and analyzed. The researchers found that dystrophin was restored to levels ranging from 3 to 90% of normal, depending on muscle type. In cardiac muscle, dystrophin levels in the dog receiving the highest dose reached 92% of normal. The treated dogs also showed improved muscle structure.
Other researchers are studying gene therapy to inactivate the offending gene (in this case the "exon 51") and restoring the expression of dystrophin. This is called antisense therapy. In a 2010 study, the researchers reported that "Antisense oligonucleotide-mediated skipping of exon 51 in cultured myoblasts from an affected [cavalier] dog restored the reading frame and protein expression". Translation: that is good, promising news.
For now, however, the prognosis is that affected cavaliers will not survive very long after severe symptoms appear. The lack of dystrophin eventually will begin to affect the contractibility of the dog’s heart.
RETURN TO TOP
Breeders’ Responsibilities
This form of muscular dystrophy is an hereditary disease passed by at least parent to its litters. Carriers can be identified. See DNA Testing, above.
RETURN TO TOP
Research News
August 2018:
CRISPR allows the mutated gene causing Duchenne muscular
dystrophy to restore dystrophin expression in four CKCS-mixes.
 In
an
August 2018 article, a team of researchers from Dallas and the UK
(Leonela Amoasii, John C.W. Hildyard, Hui Li, Efrain Sanchez-Ortiz, Alex
Mireault, Daniel Caballero, Rachel Harron, Thaleia-Rengina Stathopoulou,
Claire Massey, John M. Shelton, Rhonda Bassel-Duby, Richard J. Piercy,
Eric N. Olson [right]) tested the value of gene editing the
deltaE50-MD (ΔEx50) mutated gene in four one-month old cavalier King
Charles spaniel mix puppies (Bichon Frise x CKCS female mated with male
Beagles). Two of the puppies were injected with adeno-associated viruses
to deliver CRISPR gene editing components intra-muscularly and the other
two were injected systemically in the cephalic vein. The four dogs were
euthanized six to eight weeks later, and muscle tissue was collected and
analyzed. The researchers found that dystrophin was restored to levels
ranging from 3 to 90% of normal, depending on muscle type. In cardiac
muscle, dystrophin levels in the dog receiving the highest dose reached
92% of normal. The treated dogs also showed improved muscle structure.
In
an
August 2018 article, a team of researchers from Dallas and the UK
(Leonela Amoasii, John C.W. Hildyard, Hui Li, Efrain Sanchez-Ortiz, Alex
Mireault, Daniel Caballero, Rachel Harron, Thaleia-Rengina Stathopoulou,
Claire Massey, John M. Shelton, Rhonda Bassel-Duby, Richard J. Piercy,
Eric N. Olson [right]) tested the value of gene editing the
deltaE50-MD (ΔEx50) mutated gene in four one-month old cavalier King
Charles spaniel mix puppies (Bichon Frise x CKCS female mated with male
Beagles). Two of the puppies were injected with adeno-associated viruses
to deliver CRISPR gene editing components intra-muscularly and the other
two were injected systemically in the cephalic vein. The four dogs were
euthanized six to eight weeks later, and muscle tissue was collected and
analyzed. The researchers found that dystrophin was restored to levels
ranging from 3 to 90% of normal, depending on muscle type. In cardiac
muscle, dystrophin levels in the dog receiving the highest dose reached
92% of normal. The treated dogs also showed improved muscle structure.
December 2016: Whole genome, next generation sequencing detects new muscular dystrophy mutation in a cavalier. In a December 2016 article, a team of USA and Italian researchers (Nghiem PP, Bello L, Balog-Alvarez C, López SM, Bettis A, Barnett H, Hernandez B, Schatzberg SJ, Piercy RJ, Kornegay JN) applied "whole genome, next generation sequencing" (WGS) to evaluate a cavalier King Charles spaniel affected with muscular dystrophy (DMD). WGS detected a deletion mutation in a DMD gene hotspot which is distinct from a previously identified mutation.
EDITOR'S NOTE: Unfortunately, it appears that the cavalier in this study was being used as a laboratory dog -- much like a "lab rat" or "lab Beagle" -- for nearly five years. The researchers confess in the article that after the dog, named Buckley, was diagnosed with DMD, its owner "donated" Buckley "for further study". It apparently was passed from the University of Georgia veterinary school to the University of North Carolina - Chapel Hill medical school to the Texas A&M veterinary school, all employment venues described on the curriculum vitae of one of the researchers, Dr. Joe Kornegay. Buckley died after five years in captivity "due to complications during anesthesia", presumably in preparation for yet another experimental surgical procedure. If you don't believe us, here are two quotes about Buckley in the article itself, along with his photograph:
"A 6-month-old, intact male CKCS dog (named Buckley) was presented to the University of Georgia Small Animal Teaching Hospital for a 3 months history of dysphagia, ptyalism, nasal congestion, non-progressive coughing, lethargy, and decreased activity compared to other household dogs. The dog was obtained from a CKCS dog breeder at 3 months of age and noted to be smaller and have a weak bark compared to its littermates."
"Due to the dog’s condition, an inherited muscle disease was suspected, and it was donated for further study. The dog was subsequently transferred to the University of North Carolina-Chapel Hill and then to Texas A&M University. Gradual disease progression was observed over the next 5 years, at which time the dog died due to complications during anesthesia."
August 2011: Cavaliers' DNA needed for muscular dystrophy test validation. VetNostic Laboratories needs 50 cavalier King Charles spaniels to contribute DNA samples to validate a test for dystrophin-deficient muscular dystrophy in the breed (CKCS-MD). Collection is performed using a cheek swab kit which consists of three swabs per dog. A self addressed, postage paid, return envelope will be provided. Once the lab receives the swabs, it will take about a week to obtain the results. Testing is free and result certificates will be provided. To obtain a collection kit along with instructions, please email Rob Mason rmason@mdlab.com or call 877-255-9208. Website www.vetnostic.com
November 2004:
Cavaliers diagnosed with MD plus myalgia and myoglobinuria.
 In
a November 2004
abstract, researchers Scott J. Schatzberg and G. Diane Shelton
(right) reported diagnosing dystrophin deficient muscular dystrophy
in three unrelated cavalier King Charles spaniels. The dogs were
initially evaluated at age 4 to 5 months for stiff hunched-up gaits and
myalgia (painful or tender muscles). Serum creatine kinase (CK) levels
were very high, ranging from 83,000 to 358,000 IU/L (normal: 50–275
IU/L). Myoglobinuria (the presence of myoglobin in the urine, usually
associated with muscle destruction) was found in two of the dogs. The
absence of dystrophin was confirmed.
In
a November 2004
abstract, researchers Scott J. Schatzberg and G. Diane Shelton
(right) reported diagnosing dystrophin deficient muscular dystrophy
in three unrelated cavalier King Charles spaniels. The dogs were
initially evaluated at age 4 to 5 months for stiff hunched-up gaits and
myalgia (painful or tender muscles). Serum creatine kinase (CK) levels
were very high, ranging from 83,000 to 358,000 IU/L (normal: 50–275
IU/L). Myoglobinuria (the presence of myoglobin in the urine, usually
associated with muscle destruction) was found in two of the dogs. The
absence of dystrophin was confirmed.
RETURN TO TOP
Related Links
RETURN TO TOP
Veterinary Resources
An unusual myopathy in a dog. B. R. Jones, A.C. Johnstone. New
Zealand Vet. J. 1982;30:119-121. Quote: A nine-month-old Cavalier King
Charles Spaniel (right) was examined because it could not withdraw its tongue
into its mouth and had difficulty eating and drinking. These clinical
signs were first observed when the dog was two months of age. On
percussion of the tongue a dimple could be produced and there was
electromyographic evidence of myotonia. Histological examination showed
replacement of muscle fibres by adipose tissue; focal areas of
myonecrosis, neutrophil infiltration and proliferation of
sub-sarcolemmal nuclei. These changes were considered to be consistent
with a primary myopathy similar to paramyotonia in man. No related dogs
were found to be affected.
Newly identified neuromuscular disorders. Scott J. Schatzberg, G. Diane Shelton. Vet. Clin. Small Anim. November 2004; doi: 10.1016/j.cvsm.2004.06.001. Quote: Dystrophinopathy in Cavalier King Charles Spaniels: Three unrelated male Cavalier King Charles Spaniels were diagnosed recently with dystrophin deficient MD (G.D. Shelton, DVM, PhD, unpublished data, 2004). The dogs were initially evaluated at 4 to 5 months of age for a stiff hunched-up gait and myalgia. Serum CK levels were elevated markedly and persistently, ranging from 83,000 to 358,000 IU/L (normal: 50–275 IU/L). Myoglobinuria was documented in two dogs. Histologic examination of muscle biopsy specimens demonstrated the dystrophic phenotype, and immunohistochemical staining confirmed the absence of dystrophin. The clinical presentations of myalgia and myoglobinuria in these dogs varied somewhat from previously described cases of dystrophinopathy. Mutational analyses have not been performed in any of the dogs.
Efficient mapping of mendelian traits in dogs through genome-wide association. Elinor K Karlsson, Izabella Baranowska, Claire M Wade, Nicolette H C Salmon Hillbertz, Michael C Zody, Nathan Anderson, Tara M Biagi, Nick Patterson, Gerli Rosengren Pielberg, Edward J Kulbokas III, Kenine E Comstock, Evan T Keller, Jill P Mesirov, Henrik von Euler, Olle Kämpe, Åke Hedhammar, Eric S Lander, Göran Andersson, Leif Andersson, Kerstin Lindblad-Toh. Nature Genetics. Sept. 2007;39:1321 - 1328. Quote: "With several hundred genetic diseases and an advantageous genome structure, dogs are ideal for mapping genes that cause disease. Here we report the development of a genotyping array with ~27,000 SNPs and show that genome-wide association mapping of mendelian traits in dog breeds can be achieved with only ~20 dogs. Specifically, we map two traits with mendelian inheritance: the major white spotting (S) locus and the hair ridge in Rhodesian ridgebacks. For both traits, we map the loci to discrete regions of <1 Mb. Fine-mapping of the S locus in two breeds refines the localization to a region of ~100 kb contained within the pigmentation-related gene MITF. Complete sequencing of the white and solid haplotypes identifies candidate regulatory mutations in the melanocyte-specific promoter of MITF. Our results show that genome-wide association mapping within dog breeds, followed by fine-mapping across multiple breeds, will be highly efficient and generally applicable to trait mapping, providing insights into canine and human health."
Muscular dystrophy in Cavalier King Charles spaniels. Piercy, Richard. J. and Walmsley, Gemma. Vet Rec. 2009 165 (2), p. 62. Quote: We have recently identified the genetic cause of a form of muscular dystrophy in CKCS. The causative mutation is in the dystrophin gene and the X-linked disease is associated with weakness, muscle atrophy and exercise intolerance, detectable from a few months of age. Prominent signs in affected dogs are dysphagia [the symptom of difficulty in swallowing] and macroglossia (enlarged tongue)[tongue enlargement that leads to functional and cosmetic problems]. Serum creatine kinase is usually markedly elevated. Male dogs with the mutation [are] clinically affected and female dogs with the mutation are silent carriers. We are also keen to hear from veterinary surgeons who believe they may have seen an affected dog in their practice, in order to estimate the prevalence of this disease and limit its spread by genetic testing.
Mutational Analysis of Dystrophin-deficient Muscular Dystrophy in Cavalier King Charles Spaniels. G. L. Walmsley, V. Arechavala-Gomeza, M. Fernandez-Fuente, N. Nagel, R. Stanley, K. Chandler, F. Muntoni, G. D. Shelton, and R.J. Piercy. J Vet Intern Med. 2009; 23(3):741 (ACVIM 27th Ann. Vet. Med. Forum Abstract Program: Abstract 189). Quote: "Canine dystrophin-deficient muscular dystrophy, analogous to Duchenne muscular dystrophy of humans, is a severe inherited degenerative disorder of striated muscle. This debilitating and ultimately fatal condition results in a progressive destruction of skeletal and cardiac muscle due to mutations in the gene encoding dystrophin, a structural protein that links the contractile apparatus to the sarcolemma. The disorder is seen in several canine breeds but the genetic cause has only been reported in the Golden Retriever, German Short-haired Pointer and Rottweiler. Here we present the findings of clinical, histopathological and molecular characterisation of this condition in Cavalier King Charles Spaniels. A 10 month old male neutered client-owned Cavalier King Charles Spaniel from the United Kingdom was presented with a chronic progressive history of lethargy, exercise intolerance and dysphagia. The dog was tetraparetic with poor skeletal muscle mass (body condition score 52/9), reduced spinal reflexes, macroglossia and restricted jaw movement. Investigations documented a marked elevation in creatine kinase activity (33,695U/l; 61–394U/l) and electromyography revealed spontaneous activity indicative of a primary muscle disorder (complex repetitive discharges and pseudomyotonia). Dystrophin deficient muscular dystrophy was diagnosed on the basis of skeletal muscle histopathology, immunohistochemistry and immunoblotting using monoclonal antibodies to the dystrophin rod and carboxy termini. Oligonucleotide primer pairs designed for RT-PCR to amplify overlapping regions of dystrophin cDNA identified, following sequencing, an exon deletion and a frame-shift not present in control cDNA, that is predicted to result in premature termination of the protein product. Sequencing the associated genomic DNA confirmed the causative (and novel) mutation. The ability of antisense oligonucleotide induced exon skipping to restore the reading frame was demonstrated in vitro in cultured myoblasts from the affected dog. Sequencing of amplified DNA from an additional Cavalier King Charles Spaniel with dystrophin-deficient muscular dystrophy from North America identified the same mutation. In conclusion, dystrophin deficient muscular dystrophy in the Cavalier King Charles Spaniel may provide an excellent model for Duchenne muscular dystrophy due to the potential application for trials of antisense oligonucleotide-mediated exon skipping – one of the more promising research directions for genetic therapy in this fatal disorder."
A Duchenne Muscular Dystrophy Gene Hot
Spot Mutation in Dystrophin-Deficient Cavalier King Charles Spaniels
Is Amenable to Exon 51 Skipping. Gemma L. Walmsley, Virginia
Arechavala-Gomeza, Marta Fernandez-Fuente, Margaret M. Burke, Nicole Nagel,
Angela Holder, Rachael Stanley, Kate Chandler, Stanley L. Marks, Francesco
Muntoni, G. Diane Shelton, Richard J. Piercy. J. Plos One. January 2010;
5(1):e8647. Quote: Background: Duchenne muscular dystrophy (DMD), which afflicts
1 in 3500 boys, is one of the most common genetic disorders of children. This
fatal degenerative condition is caused by an absence or deficiency of dystrophin
in striated muscle. Most affected patients have inherited or spontaneous
deletions in the dystrophin gene that disrupt the reading frame resulting in
unstable truncated products. For these patients, restoration of the reading
frame via antisense oligonucleotide-mediated exon skipping is a promising
therapeutic approach. The major DMD deletion “hot spot” is found between exons
45 and 53, and skipping exon 51 in particular is predicted to ameliorate the
dystrophic phenotype in the greatest number of patients. Currently the mdx mouse
is the most widely used animal model of DMD, although its mild phenotype limits
its suitability in clinical trials. The Golden Retriever muscular dystrophy
(GRMD) model has a severe phenotype, but due to its large size, is expensive to
use. Both these models have mutations in regions of the dystrophin gene distant
from the commonly mutated DMD “hot spot”. Methodology/Principal Findings: Here
we describe the severe phenotype, histopathological findings, and molecular
analysis of
Cavalier King Charles Spaniels with
dystrophin-deficient muscular dystrophy (CKCS-MD).
... Here we describe a spontaneous canine model of DMD in the
Cavalier King Charles Spaniel (CKCS), a toy breed,
specifically bred for its small size (5– 8 kg) and amiable
temperament. In this manuscript we report the clinical, pathological
and molecular characterisation of the model and investigate the
suitability of exon skipping-mediated therapy. In this study we
sought to determine the genetic basis of dystrophin-deficient
muscular dystrophy in 3 CKCS dogs. The novel splice
site mutation of the dystrophin gene that we have identified in
these affected dogs from the UK and the USA occurs in a region that
is most commonly mutated in humans, and gives rise to a single exon
deletion (exon 50) in the dystrophin mRNA. ...
 The index case
was a 10-month-old male neutered client-owned CKCS
(see photo)
from the United Kingdom that was presented to the Royal Veterinary
College for investigation of a generalised neuromuscular disorder.
The dog had been in the owners’ possession from 2 months of age and
the puppy had always been of small stature, poorly muscled and
exercise intolerant; the main presenting complaint, however, was a 3
month progressive history of dysphagia. ... Two additional young
male CKCS from the USA were also diagnosed with
dystrophin deficient muscular dystrophy (dogs 2 and 3). Dog 2 was a 6-month-old
intact male that was presented to a general veterinarian because of dysphagia
and decreased range of jaw motion, first noted by the owner at 2 months of age.
A marked elevation in serum CK activity (64,918 U/L) was measured. Dog 3 was an
8-month-old neutered male that was presented to the Veterinary Teaching
Hospital, University of California, Davis, with a history of gagging when eating
and drinking since acquired as a 6-week-old puppy, combined with exercise
intolerance. On physical examination, restricted jaw mobility and macroglossia
were noted. The serum CK activity was markedly elevated (58,508 U/L). Thoracic
radiography revealed a normal shape and size to the cardiac silhouette and
echocardiography revealed very mild mitral and tricuspid regurgitation (believed
to be clinically irrelevant) and normal left ventricular function.
Electromyography revealed spontaneous repetitive discharges and pseudomyotonia
in all muscles examined. ... The dogs harbour a missense mutation in the
5′ donor splice site of exon 50 that results in deletion of exon 50 in mRNA
transcripts and a predicted premature truncation of the translated protein.
Antisense oligonucleotide-mediated skipping of exon 51 in cultured myoblasts
from an affected dog restored the reading frame and protein expression.
Conclusions/Significance: Given the small size of the breed, the amiable
temperament and the nature of the mutation, we propose that CKCS-MD is a
valuable new model for clinical trials of antisense oligonucleotide-induced exon
skipping and other therapeutic approaches for DMD. ... Further work, ideally
utilizing a colony of CKCS-MD dogs, would enable comprehensive phenotypic
characterization of various organ systems, including the heart. Furthermore, if
the cardiac muscle changes are found to be comparatively mild in this model, it
would provide an opportunity to evaluate factors contributing to this phenotype.
The index case
was a 10-month-old male neutered client-owned CKCS
(see photo)
from the United Kingdom that was presented to the Royal Veterinary
College for investigation of a generalised neuromuscular disorder.
The dog had been in the owners’ possession from 2 months of age and
the puppy had always been of small stature, poorly muscled and
exercise intolerant; the main presenting complaint, however, was a 3
month progressive history of dysphagia. ... Two additional young
male CKCS from the USA were also diagnosed with
dystrophin deficient muscular dystrophy (dogs 2 and 3). Dog 2 was a 6-month-old
intact male that was presented to a general veterinarian because of dysphagia
and decreased range of jaw motion, first noted by the owner at 2 months of age.
A marked elevation in serum CK activity (64,918 U/L) was measured. Dog 3 was an
8-month-old neutered male that was presented to the Veterinary Teaching
Hospital, University of California, Davis, with a history of gagging when eating
and drinking since acquired as a 6-week-old puppy, combined with exercise
intolerance. On physical examination, restricted jaw mobility and macroglossia
were noted. The serum CK activity was markedly elevated (58,508 U/L). Thoracic
radiography revealed a normal shape and size to the cardiac silhouette and
echocardiography revealed very mild mitral and tricuspid regurgitation (believed
to be clinically irrelevant) and normal left ventricular function.
Electromyography revealed spontaneous repetitive discharges and pseudomyotonia
in all muscles examined. ... The dogs harbour a missense mutation in the
5′ donor splice site of exon 50 that results in deletion of exon 50 in mRNA
transcripts and a predicted premature truncation of the translated protein.
Antisense oligonucleotide-mediated skipping of exon 51 in cultured myoblasts
from an affected dog restored the reading frame and protein expression.
Conclusions/Significance: Given the small size of the breed, the amiable
temperament and the nature of the mutation, we propose that CKCS-MD is a
valuable new model for clinical trials of antisense oligonucleotide-induced exon
skipping and other therapeutic approaches for DMD. ... Further work, ideally
utilizing a colony of CKCS-MD dogs, would enable comprehensive phenotypic
characterization of various organ systems, including the heart. Furthermore, if
the cardiac muscle changes are found to be comparatively mild in this model, it
would provide an opportunity to evaluate factors contributing to this phenotype.
Mammalian Models of Duchenne Muscular Dystrophy: Pathological Characteristics and Therapeutic Applications. Akinori Nakamura and Shin'ichi Takeda. J.Biomed.Biotechnol.; Jan 2011; 2011:184393. Quote: "5.3. Cavalier King Charles Spaniels with Muscular Dystrophy (CKCS-MD) -- 5.3.1. Pathological Characteristics -- Very recently, it was reported that another dystrophic dog, the Cavalier King Charles Spaniel with dystrophin-deficient muscular dystrophy (CKCS-MD) has a severe phenotype. This canine model has a missense mutation in the 5′ donor splice site of exon 50 resulting in deletion of exon 50 in mRNA transcripts and a predicted premature truncation of the translated protein. 5.3.2. Therapeutic Applications -- The therapeutic strategy of exon skipping in the GRMD or CXMDJ models provides a significant advantage in clinical trials, but also has the unfavorable characteristic that the disease-causing mutation does not include the region of the DMD gene that is most commonly mutated in human DMD patients. About 60% of DMD patients harbor deletions in exons 45–55 of the DMD gene [55, 56]. As described previously, the exon 51 skipping technique, which is being employed in clinical trials [30, 31], is applicable to DMD patients which have deletions of exon 50, exon 52, exons 48–50, or exons 49-50, among others, and is feasible for more patients than other exon skipping strategies [27, 57, 58]. AO-mediated skipping of exon 51 in cultured myoblasts of the CKCS-MD dog restored the reading frame and protein expression [17]. This observation suggests that the use of this canine model would be valuable in the preclinical trials of therapy based upon exon 51 skipping."
Genetic Connection: A Guide to Health Problems in Purebred Dogs, Second Edition. Lowell Ackerman. July 2011; AAHA Press; pg 127. Quote: "Cavalier King Charles spaniels ...are susceptible to an X-linked myopathy similar to Duchenne-type muscular dystrophy in humans. ... The defect has been characterized in the Cavalier King Charles spaniel ... and so it is likely that other genetic tests will eventually become available."
Canine models of Duchenne muscular dystrophy and their use in therapeutic strategies. Joe N. Kornegay, Janet R. Bogan, Daniel J. Bogan, Martin K. Childers, Juan Li, Peter Nghiem, David A. Detwiler, C. Aaron Larsen, Robert W. Grange, Ratna K. Bhavaraju-Sanka, Sandra Tou, Bruce P. Keene, James F. Howard Jr., Jiahui Wang, Zheng Fan, Scott J. Schatzberg, Martin A. Styner, Kevin M. Flanigan, Xiao Xiao, Eric P. Hoffman. Mamm. Genome. Jan. 2012;23(1):85-108. Quote: "The progression of clinical disease and serum creatine kinase (CK) levels in canine X-linked muscular dystrophy (CXMD) was studied in 7 dogs from birth to 12-14 months and in 18 dogs at varying intervals from birth to 8 weeks. One affected male was studied from age 3.5 to 6 years, and all pups were descendants of this dog. A lethal neonatal form was recognized in some pups. In the more typical form, clinical signs of stunting, weakness and gait abnormalities were evident by 6-9 weeks and were progressive, leading to marked muscle atrophy, fibrosis and contractures by 6 months. Serum CK levels were markedly elevated, such that affected pups could be identified by 1 week. CK values increased until 6-8 weeks, then plateaued at approx. 100 times normal. Affected females and beagle-cross dogs were less severely affected than large breed-cross dogs. In the 2 adult dogs with cardiac insufficiency CK levels had decreased to 5-15 times normal. These studies show that CXMD and Duchenne muscular dystrophy have striking phenotypic as well as genotypic similarities. In addition, these studies of CXMD suggest that in females and in smaller dogs the same genetic defect results in a less severe clinical disease."
Inhibition of CD26/DPP-IV enhances donor muscle cell engraftment and stimulates sustained donor cell proliferation. Maura H Parker, Carol Loretz, Ashlee E Tyler, Lauren Snider, Rainer Storb, Stephen J Tapscott. Skelet Muscle. 2012; 2: 4. Published online 2012 February 16. doi: 10.1186/2044-5040-2-4 Quote: "Background: Transplantation of myogenic stem cells possesses great potential for long-term repair of dystrophic muscle. In murine-to-murine transplantation experiments, CXCR4 expression marks a population of adult murine satellite cells with robust engraftment potential in mdx mice, and CXCR4-positive murine muscle-derived SP cells home more effectively to dystrophic muscle after intra-arterial delivery in mdx5cv mice. Together, these data suggest that CXCR4 plays an important role in donor cell engraftment. Therefore, we sought to translate these results to a clinically relevant canine-to-canine allogeneic transplant model for Duchenne muscular dystrophy (DMD) and determine if CXCR4 is important for donor cell engraftment. Methods: In this study, we used a canine-to-murine xenotransplantation model to quantitatively compare canine muscle cell engraftment, and test the most effective cell population and modulating factor in a canine model of DMD using allogeneic transplantation experiments. Results: We show that CXCR4 expressing cells are important for donor muscle cell engraftment, yet FACS sorted CXCR4-positive cells display decreased engraftment efficiency. However, diprotin A, a positive modulator of CXCR4-SDF-1 binding, significantly enhanced engraftment and stimulated sustained proliferation of donor cells in vivo. Furthermore, the canine-to-murine xenotransplantation model accurately predicted results in canine-to-canine muscle cell transplantation. Conclusions: Therefore, these results establish the efficacy of diprotin A in stimulating muscle cell engraftment, and highlight the pre-clinical utility of a xenotransplantation model in assessing the relative efficacy of muscle stem cell populations."
Cardiac structure and function in female carriers of a canine model of Duchenne muscular dystrophy. A.M. Kane, T.C. DeFrancesco, M.C. Boyle, D.E. Malarkey, J.W. Ritchey, C.E. Atkins, J.M. Cullen, J.N. Kornegay, B.W. Keene. Research in Vet. Sci. Dec. 2012. Quote: "This investigation tested the hypothesis that carriers of golden retriever muscular dystrophy (GRMD), a genetically homologous condition of Duchenne muscular dystrophy (DMD), have quantifiable abnormalities in myocardial function, structure, or cardiac rhythm. Eleven GRMD carriers and four matched controls had cardiac evaluations and postmortem examinations. 24-h ECG Holter monitoring disclosed ventricular ectopy in 10 of 11 carriers and 2 of 4 controls. Conventional echocardiography failed to demonstrate significant differences between carriers and controls in systolic function. All carriers had multifocal, minimal to marked myofiber necrosis, fibrosis, mineralization, inflammation, and/or fatty change in their hearts. Immunohistochemistry revealed a mosaic dystrophin deficiency in scattered cardiac myofibers in all carriers. No controls had cardiac histologic lesions; all had uniform dystrophin staining. Despite cardiac mosaic dystrophin expression and degenerative cardiac lesions, GRMD carriers at up to 3 years of age could not be distinguished statistically from normal controls by echocardiography or 24-h Holter monitoring."
Duchenne muscular dystrophy gene therapy in the canine model. Dongsheng Duan. Human Gene Therapy Clinical Development. January 2015. doi:10.1089/hum.2015.006. Quote: "Duchenne muscular dystrophy (DMD) is an X-linked lethal muscle disease caused by dystrophin deficiency. Gene therapy has significantly improved the outcome of dystrophin-deficient mice. Yet, clinical translation has not resulted in the expected benefits in human patients. This translational gap is largely due to the insufficient modeling of DMD in mice. Specifically, mice lacking dystrophin show minimum dystrophic symptoms and they do not respond to the gene therapy vector in the same way as human patients do. Further, the size of a mouse is hundredfolds smaller than a boy, making it impossible to scale-up gene therapy in a mouse model. None of these limitations exist in the canine DMD (cDMD) model. For this reason, cDMD dogs have been considered a highly valuable platform to test experimental DMD gene therapy. Over the last three decades, a variety of gene therapy approaches have been evaluated in cDMD dogs using a number of non-viral and viral vectors. These studies have provided critical insight for the development of an effective gene therapy protocol in human patients. This review discusses the history, current status and future directions of the DMD gene therapy in the canine model."
Animal models of Duchenne muscular dystrophy: from basic mechanisms to gene therapy. Joe W. McGreevy, Chady H. Hakim, Mark A. McIntosh, Dongsheng Duan. Disease Models & Mechanisms. March 2015;8(3):195–213. Quote: "Duchenne muscular dystrophy (DMD) is a progressive muscle-wasting disorder. It is caused by loss-of-function mutations in the dystrophin gene. Currently, there is no cure. A highly promising therapeutic strategy is to replace or repair the defective dystrophin gene by gene therapy. Numerous animal models of DMD have been developed over the last 30 years, ranging from invertebrate to large mammalian models. mdx mice are the most commonly employed models in DMD research and have been used to lay the groundwork for DMD gene therapy. After ~30 years of development, the field has reached the stage at which the results in mdx mice can be validated and scaled-up in symptomatic large animals. The canine DMD (cDMD) model will be excellent for these studies. In this article, we review the animal models for DMD, the pros and cons of each model system, and the history and progress of preclinical DMD gene therapy research in the animal models. We also discuss the current and emerging challenges in this field and ways to address these challenges using animal models, in particular cDMD dogs. ... Dystrophin gene mutations have been mapped in nine cDMD breeds (although only four mutations have been published in peer-reviewed research articles). Point mutations have been found in the Cavalier King Charles spaniel muscular dystrophy dogs (CKCS-MD, intron 50), golden retriever muscular dystrophy dogs (GRMD, intron 6) and Rottweiler muscular dystrophy dogs (exon 52). Deletion mutations have been found in three breeds, including a small four-nucleotide deletion in exon 65 (Cocker spaniel), an exon 8–29 deletion in the Tibetan terrier, and a whole-gene deletion in the German shorthaired pointer. "
Whole genome sequencing reveals a 7 base-pair deletion in DMD exon 42 in
a dog with muscular dystrophy. Nghiem PP, Bello L,
Balog-Alvarez C, López SM, Bettis A, Barnett H, Hernandez B, Schatzberg
SJ, Piercy RJ, Kornegay JN. Mamm Genome. December 2016. Quote:
Dystrophin is a key cytoskeletal protein coded by the Duchenne muscular
dystrophy (DMD) gene located on the X-chromosome. Truncating mutations
in the DMD gene cause loss of dystrophin and the classical DMD clinical
syndrome. Spontaneous DMD gene mutations and associated phenotypes occur
in several other species.
 The mdx mouse model and the golden retriever
muscular dystrophy (GRMD) canine model have been used extensively to
study DMD disease pathogenesis and show efficacy and side effects of
putative treatments. Certain DMD gene mutations in high-risk, the
so-called hot spot areas can be particularly helpful in modeling
molecular therapies. Identification of specific mutations has been
greatly enhanced by new genomic methods. Whole genome, next generation
sequencing (WGS) has been recently used to define DMD patient mutations,
but has not been used in dystrophic dogs. A dystrophin-deficient
Cavalier King Charles Spaniel (CKCS) dog was evaluated at the
functional, histopathological, biochemical, and molecular level. ... A
6-month-old, intact male CKCS dog (named Buckley) was
presented to the University of Georgia Small Animal Teaching Hospital
for a 3 months history of dysphagia, ptyalism, nasal congestion,
non-progressive coughing, lethargy, and decreased activity compared to
other household dogs. The dog was obtained from a CKCS
dog breeder at 3 months of age and noted to be smaller and have a weak
bark compared to its littermates. ... Due to the dog’s condition, an
inherited muscle disease was suspected, and it was donated for further
study. The dog was subsequently transferred to the University of North
Carolina-Chapel Hill and then to Texas A&M University. Gradual disease
progression was observed over the next 5 years, at which time the dog
died due to complications during anesthesia. ... The
affected dog's phenotype was compared to the previously reported canine
dystrophinopathies. WGS was then used to detect a 7 base pair deletion
in DMD exon 42 (c.6051-6057delTCTCAAT mRNA), predicting a frameshift in
gene transcription and truncation of dystrophin protein translation. The
deletion was confirmed with conventional PCR and Sanger sequencing. This
mutation is in a secondary DMD gene hotspot area distinct from the one
identified earlier at the 5' donor splice site of intron 50 in the CKCS
breed. ...
The mdx mouse model and the golden retriever
muscular dystrophy (GRMD) canine model have been used extensively to
study DMD disease pathogenesis and show efficacy and side effects of
putative treatments. Certain DMD gene mutations in high-risk, the
so-called hot spot areas can be particularly helpful in modeling
molecular therapies. Identification of specific mutations has been
greatly enhanced by new genomic methods. Whole genome, next generation
sequencing (WGS) has been recently used to define DMD patient mutations,
but has not been used in dystrophic dogs. A dystrophin-deficient
Cavalier King Charles Spaniel (CKCS) dog was evaluated at the
functional, histopathological, biochemical, and molecular level. ... A
6-month-old, intact male CKCS dog (named Buckley) was
presented to the University of Georgia Small Animal Teaching Hospital
for a 3 months history of dysphagia, ptyalism, nasal congestion,
non-progressive coughing, lethargy, and decreased activity compared to
other household dogs. The dog was obtained from a CKCS
dog breeder at 3 months of age and noted to be smaller and have a weak
bark compared to its littermates. ... Due to the dog’s condition, an
inherited muscle disease was suspected, and it was donated for further
study. The dog was subsequently transferred to the University of North
Carolina-Chapel Hill and then to Texas A&M University. Gradual disease
progression was observed over the next 5 years, at which time the dog
died due to complications during anesthesia. ... The
affected dog's phenotype was compared to the previously reported canine
dystrophinopathies. WGS was then used to detect a 7 base pair deletion
in DMD exon 42 (c.6051-6057delTCTCAAT mRNA), predicting a frameshift in
gene transcription and truncation of dystrophin protein translation. The
deletion was confirmed with conventional PCR and Sanger sequencing. This
mutation is in a secondary DMD gene hotspot area distinct from the one
identified earlier at the 5' donor splice site of intron 50 in the CKCS
breed. ...
Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Leonela Amoasii, John C.W. Hildyard, Hui Li, Efrain Sanchez-Ortiz, Alex Mireault, Daniel Caballero, Rachel Harron, Thaleia-Rengina Stathopoulou, Claire Massey, John M. Shelton, Rhonda Bassel-Duby, Richard J. Piercy, Eric N. Olson. Science. August 2018;doi: 10.1126/aau1549. Quote: Mutations in the gene encoding dystrophin, a protein that maintains muscle integrity and function, cause Duchenne muscular dystrophy (DMD). ... The mutation carried by the deltaE50-MD canine model of DMD leads to loss of exon 50 and moreover can be corrected by skipping of exon 51, making this a valuable model for translational studies. First identified as a naturally occurring, spontaneous mutation in Cavalier King Charles Spaniels and now maintained on a beagle background, this model (in contrast to mice) exhibits many of the clinical and pathological features of the human disease, such as muscle weakness, atrophy and fibrosis. ... The deltaE50-MD dog model of DMD harbors a mutation corresponding to a mutational “hot spot” in the human DMD gene. We used adeno-associated viruses to deliver CRISPR gene editing components to four dogs and examined dystrophin protein expression 6 weeks after intramuscular delivery (n=2) or 8 weeks after systemic delivery (n=2). ... Carrier female Beagle (RCC strain)-cross (F3 generation) dogs derived from an original founder Bichon-Frise cross Cavalier King Charles Spaniel female were mated with male Beagles (RCC strain) to produce offspring. ... After systemic delivery in skeletal muscle, dystrophin was restored to levels ranging from 3 to 90% of normal, depending on muscle type. In cardiac muscle, dystrophin levels in the dog receiving the highest dose reached 92% of normal. The treated dogs also showed improved muscle histology. These large animal data support the concept that, with further development, gene editing approaches may prove clinically useful for the treatment of DMD.
CONNECT WITH US